August 31, 2021
by Michael Black
Carcinogenesis, or the process of transformation of cells to a cancerous state, often is associated with aberrations in messenger RNA splicing. Removal of introns in pre-mRNA is an essential step in normal cell processing of mRNA prior to transcription of a peptide from a final mRNA transcript. Alternative splicing of pre-mRNA gives cells the ability to regulate multiple final mRNA products from a single gene, adding diversity to the population of protein types and sub-types that drive normal cell processes. However, abnormal perturbations in the cellular machinery responsible for alternative splicing is common in cancers and appears to be a driving mechanism for carcinogenesis (El Marabti and Younis, 2018; Ladomery, 2013).
Exitrons (exonic introns: defined as introns within protein coding exons) represent a recently discovered form of post translational modification of protein coding genes (Marquez et al., 2015; Staiger and Simpson, 2015). While exitrons are introns, they display many exon-like properties. Alternative splicing events involving exitrons add to the complexity of the proteome and thus enhance phenotypic diversity. Exitrons thus represent a novel mechanism by which perturbations in normal protein diversity may be introduced, leading to carcinogenesis or other aberrant cellular processes.
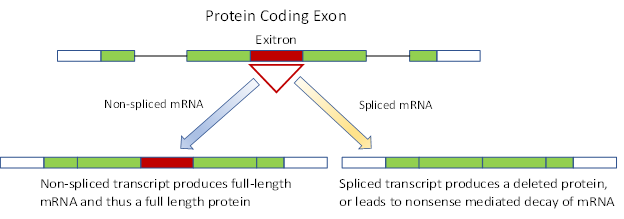
Figure 1. An exitron is an internal part of a protein coding exon and represents possible sites for alternative splicing events in gene transcription. When un-spliced, the exon produces a final full length translated protein. Exitron splicing events produce a deleted final protein product with a frameshift modified C-terminus. Alternatively the modified transcript, if it includes a premature termination codon, may result in non-sense mediated RNA decay of the transcript prior to translation.
The role of exitrons is only beginning to be understood but they add yet another level of complexity to understanding genomic and proteomic diversity. A recent in-depth analyses of exitron splicing amongst genes involved in carcinogenesis revealed exitron splicing as key events involving numerous cancer driver genes (Wang et al., 2021).
Wang et al identified 129,000 exitrons amongst 40,000 exons associated with 33 cancer types. They identified that exitrons have several key distinguishing features, such as high guanine/cytosine content, relatively short length and were most abundant in medium size exons (exons in the range of 100 to 1000 base pairs in length). The incidence of exitron splicing in cancer tissues was also much greater (63%) in tumor cells than in normal human tissue cells (17%), with the greatest incidence of exitron splicing events observed in tumors from ovary, esophageal and stomach tissues as well as acute myeloid leukemia. Differential expression of splicing factors (the genes coding proteins which function in the removal of introns in mRNA so that a final translatable product of connected exons is formed) as well as genes for proteins involved in RNA transport and mRNA repair largely explained the exitron activity in cancer versus normal human tissue.i
Several significantly exitron-spliced genes were identified in tumor tissue. These include enrichment of genes for proteins involved in regulating transcription (TAF15, FUS, EWSR1) with evidence these exitron-spliced genes may be involved in promoting progression of cancer. Another example was found in the gene encoding forkhead box protein O4 (FOXO4), where a frameshift exitron-splicing event in exon 2 was greatly up-regulated in tumor cell expression relative to normal human tissue expression. FOXO4 is involved in the regulation of cell growth and cellular differentiation, and the exon 2 mutation likely leads to loss of function of the resulting protein.
Tissue specific analyses of cancer genes showed another gene, NEFH (which encodes the neurofilament heavy chain protein) was an exitron-spliced gene associated with prostate cancer. However, in this instance, NEFH shows a much higher level of expression in normal tissues than it did in prostate tumor tissue indicating the association of this exitron-spliced gene with metastatic prostatic cancer involves down-regulation of the NEFH gene. A specific exitron-spliced variant was identified (an in-frame mutation resulting in the loss of 41 amino acids at the C-terminus of the translated protein) that, when down-regulated, leads to increased cell growth and reduction in apoptosis events. This represents the first evidence that exitron splicing events may enable tumor growth by functionally disabling normal tumor suppressor genes.
Exitron splicing adds yet another level of complexity in understanding the process of carcinogenesis and tumorous cell growth. Both the up-regulation and down-regulation of genes which experience exitron splicing events are implicated in several types of tumor cells. By implication, exitron splicing events may play key roles in other types of aberrant cell biology. Additionally, the discovery of exitron splicing places further emphasis on understanding the role of RNA splicing factors and the effects of dysregulation of their normal cellular expression levels (Cerasuolo et al., 2020).
The analyses presented in Wang et al., 2021 also demonstrates the potential of next-generation sequencing technologies and comparative computational analyses of RNA-Seq and DNA (genomic) sequence data to derive detailed insights into cellular mechanisms of action of adverse outcomes. Analyses like this, especially when extended into the realm of single cell and single nucleus sequencing technology could be extended to compare cellular MOA amongst classes of compounds with adverse cellular response, or to determine differences in tissue or cell type response to a given perturbation. Identifying sources of change in the transcriptome and proteome allow for ever more sophisticated analyses of cellular MOA when cells undergo change due to a perturbation.
ScitoVation’s computational toxicology group has extensive experience with analyses of differential gene expression from both microarray and RNA-Seq data, as well as deriving cellular MOAs from transcriptomic data (Andersen et al., 2017; Andersen et al., 2018; Black et al., 2014; McMullen et al., 2019). Recently we have been providing analyses for transcriptomic risk assessment and implementing evolving best practices for transcriptomic points of departure (Nault et al., 2020). Alternative splicing analyses with RNA-Seq data provides an additional method to explore transcriptomic changes and may prove insightful for determining key aspects of MOA in toxicology.
El Marabti, E., and Younis, I. (2018). The Cancer Spliceome: Reprograming of Alternative Splicing in Cancer. Front Mol Biosci 5, 80, 10.3389/fmolb.2018.00080.
Ladomery, M. (2013). Aberrant Alternative Splicing Is Another Hallmark of Cancer. International Journal of Cell Biology 2013, 463786, 10.1155/2013/463786.
Marquez, Y., Höpfler, M., Ayatollahi, Z., Barta, A., and Kalyna, M. (2015). Unmasking alternative splicing inside protein-coding exons defines exitrons and their role in proteome plasticity. Genome Res 25(7), 995-1007, 10.1101/gr.186585.114.
Staiger, D., and Simpson, G. G. (2015). Enter exitrons. Genome Biology 16(1), 136, 10.1186/s13059-015-0704-3.
Wang, T. Y., Liu, Q., Ren, Y., Alam, S. K., Wang, L., Zhu, Z., Hoeppner, L. H., Dehm, S. M., Cao, Q., and Yang, R. (2021). A pan-cancer transcriptome analysis of exitron splicing identifies novel cancer driver genes and neoepitopes. Mol Cell, 10.1016/j.molcel.2021.03.028.
Cerasuolo, A., Buonaguro, L., Buonaguro, F. M., and Tornesello, M. L. (2020). The Role of RNA Splicing Factors in Cancer: Regulation of Viral and Human Gene Expression in Human Papillomavirus-Related Cervical Cancer. Frontiers in Cell and Developmental Biology 8(474), 10.3389/fcell.2020.00474.